Introduction
This document describes how to use the Canto phenotype curation interface, and provides general and specific tips for choosing and interpreting terms from the Fission Yeast Phenotype Ontology (FYPO).
A phenotype is any observable characteristic or trait of an organism that results from the interactions between its genotype and the environment. For PomBase, Canto supports annotation of single- and multiple-allele phenotypes using FYPO terms and additional useful details such as evidence and experimental conditions.
When using FYPO terms -- or terms from any ontology -- always pay careful attention to the term definitions. They are usually more detailed, and often more informative, than the term names alone. For each annotation, ensure that the definition of the selected term accurately describes the experiment you are trying to capture, and that the results shown in the paper fit all parts of the term definition.
Using the interface
Single-allele phenotypes
A single allele is a mutation, or set of mutations, in one copy of a gene at one locus (which may be the endogenous locus or a different locus, such as a plasmid or an insertion at a non-native position). You can also annotate under- or overexpression of the wild type allele as a single "mutation". To annotate the phenotype of a single allele, click "Single allele" in the list of curation types. A box will pop up where you can add allele details:
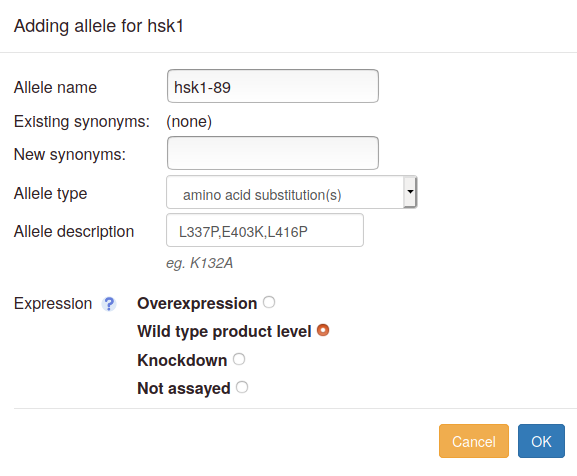
- Allele name: this field is optional. Fill this in if the allele is named, e.g. "hsk1-89". For the "wild type" and "deletion" types, a default name will be assigned. For other types, the allele will be denoted "unnamed" if no name is filled in.
- Allele name suggestions: As you type the allele name, an autocomplete pulldown may appear if there are matches to any alleles already in Canto's database. If your allele name appears, you can select it, and its type and description will be filled in. You will only have to choose the expression level. Otherwise, proceed to add allele details:
- Synonyms: Any synonyms for the allele already in the database will be listed as "Existing synonyms". You can add more in the "New synonyms" box; use commas to separate multiple entries.
-
Allele type: choose an allele type in the dropdown
menu. For some types, e.g. partial deletions or substitution,
further specifications are needed, and in these cases an "Allele
description" box appears, with an example displayed below the box
in grey text. To specify positions:
- In the description, number amino acid residues starting at the initiator methionine for all proteins including histones. It's a convention for histones that residue numbering assumes that the initiator methionine is removed, and this should be reflected in the allele name only. For example, the allele with allele name `hht1-K14A` should have a description `K15A`.
- For nonsense mutations, simply use "partial deletion, amino acid".
- For insertions, enter the one-letter symbol and number of the residue before the insertion, a hyphen, and the inserted sequence.
- For combinations of substitutions, insertions, and/or deletions, separate parts of the description with commas. For example, "A858-MKGYP,F124D" means five amino acid residues were inserted after Ala at position 858, and Phe was changed to Asp at position 124.
- For protein-coding genes, number nucleotide positions starting with the "A" of the initiator ATG, and exclude introns. (See below for how to describe changes in intron sequences.) Mutations in promoter regions can be specified by prefixing the numbers with a minus sign ("-").
- For non-coding RNA genes, number nucleotide positions starting from the annotated 5' end of the mature transcript, i.e. again exclude introns. See below for how to describe changes in intron sequences.
- For changes in intron sequences, count nucleotide residues from the starting point as described above, enter the resulting position number(s), and include the suffix "primary_transcript" (e.g. G234A-primary_transcript for a substitution, 456-555-primary_transcript for a deletion, or 188-TTCC-primary_transcript for an insertion).
- Expression level: You will be prompted to define the expression level relative to wild-type (deletion mutants are automatically set to null). Note that "expression" refers to the amount of gene product present in the assayed cells. If the product level was not measured (e.g. by Western blot for a protein), choose "Not assayed", even if a construct such as an inducible promoter was used to try to alter expression. If a wild-type allele was investigated in the absence of any other changes, then we strongly recommend setting an altered expression level.
- Descriptions for "unknown" alleles: If you know the description for any allele that is listed as "unknown" in Canto, please enter it. To do so, type in the allele name, but do not select it from the autocomplete options. Instead, proceed as if no match had been found, and you will be able to choose a type and enter a description.
Next, to find a FYPO term, type text into the search box. When suggestions from the autocomplete feature appear, choose one and proceed.
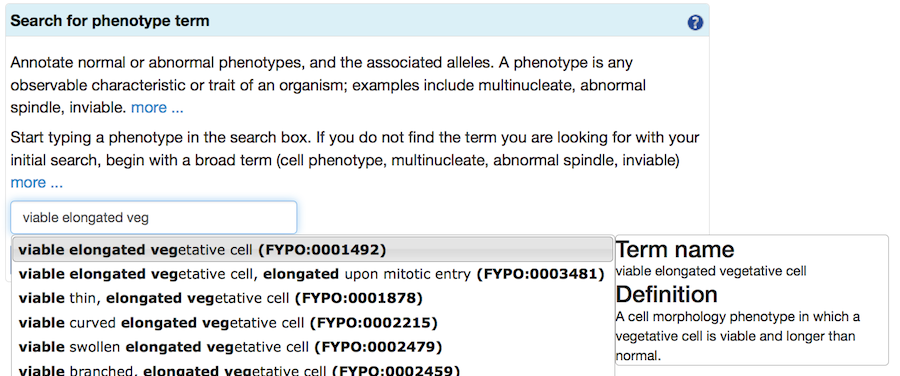
If your initial search does not find any suitable terms, try again with a broader term (some examples are provided in the specific sections below). Selecting a term takes you to a page where you can read the definition to confirm that it is applicable. More specific "child" terms will be shown (where available), and you can select one of these more specific terms in an iterative process.
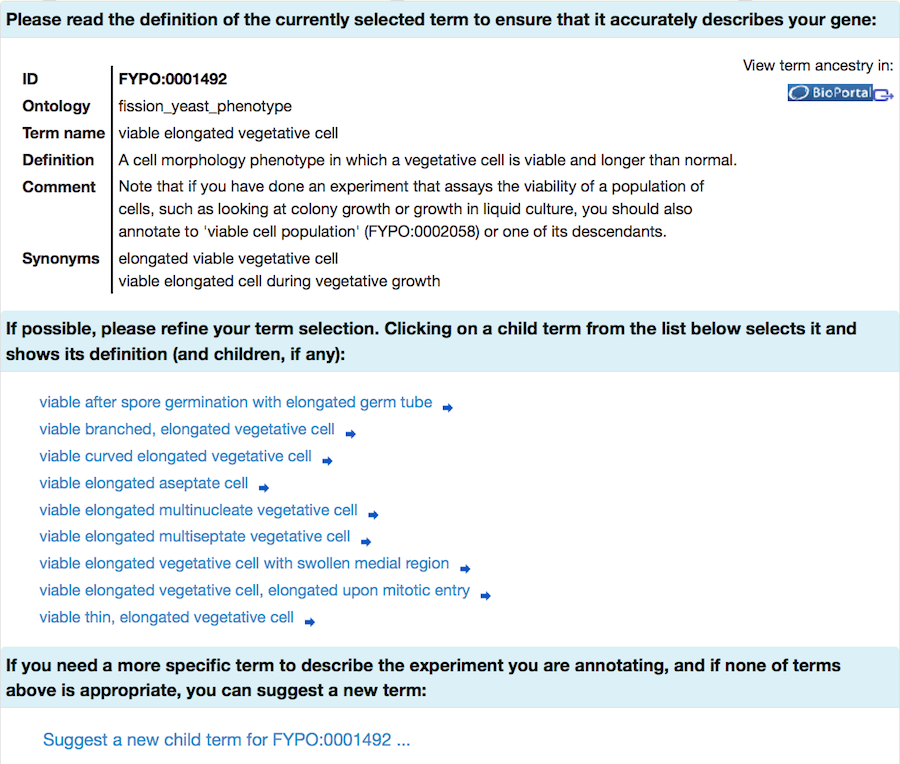
FYPO terms are organised in a hierarchical structure, and FYPO annotations should be as specific as possible to describe the data from your experiment. (More information on FYPO organisation is available on the PomBase wiki.) You will have the opportunity to request a new term if the most specific term available does not describe your gene product adequately.
After you choose a term, you will then be prompted to select evidence from a pulldown menu, and then to enter any (optional) experimental conditions. For conditions, type text and select from the autocomplete options. Several conditions can be added for one experiment. Condition terms previously used in the session appear below the text box and can be re-used by clicking on them.
Conditions are aspects of the experimental setup that may be relevant to various different methods, and are independent of what cells, strain, organism, etc. are used. Examples include:
- Glucose minimal medium vs. glucose rich medium (synonyms for some standard media are available, such as EMM or YES; carbon sources other than glucose can be specified).
- Agar plates vs. liquid medium
- Addition of certain chemicals; for instance, in a salt stress experiment it may be of interest to note what salts were added.
- Temperature (high, standard or low).
- Exclusion of some chemicals that one might normally expect to be present.
Note: you can also enter a single allele as a genotype using the genotype management interface, which is described below for multi-allele phenotypes. This is because the Chado database that Canto uses links all phenotypes to a genotype, whether that genotype specifies one or more alleles.
Multiple-allele genotypes and diploids
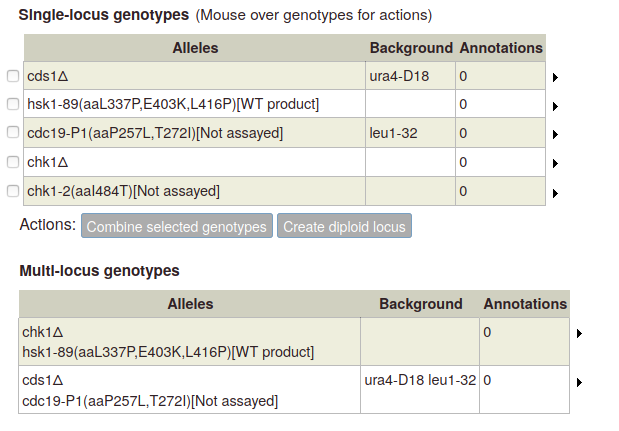
The "Genotype management" link on the paper summary page or any gene page goes to the Genotypes page, which displays genes and genotypes added for the paper. On this page you can create single- or multi-allele genotypes, and create genotypes for diploids as well as haploids.
Use the multi-allele genotype option to annotate phenotypes of a double mutant, triple mutant, or any strain in which more than one gene has its sequence or expression altered, including any case where you have more than one allele of the same gene present (e.g. one on the chromosome, and another on a plasmid). Enter details of all relevant alleles in the genotype (background details such as mating type and markers are optional).
On the left-hand side of the page is a table of genes. For each gene, you can use the "Deletion" button to add a deletion as a single-allele genotype with one click, or click "Other genotype" to bring up the allele details popup (described above).

As you add alleles, they will appear in a table on the right (any single alleles added via the "single allele phenotype" gene page option will also appear):
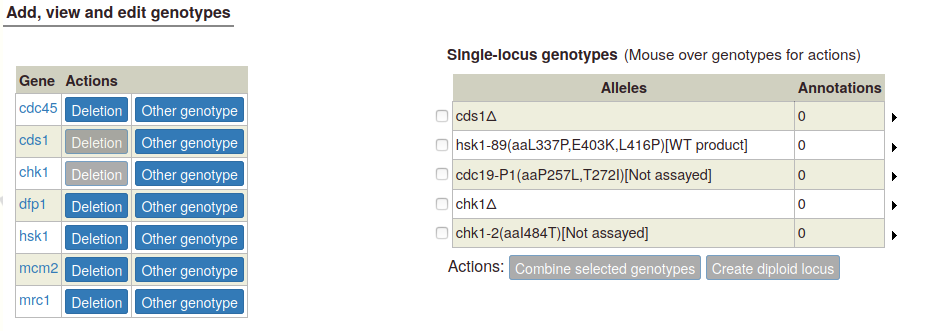
To create a diploid genotype, select one allele in the single-locus genotype table. The "Create diploid locus" button becomes enabled:
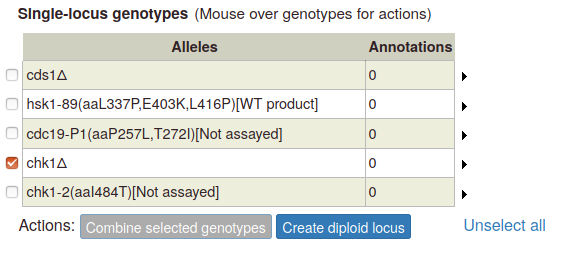
A pop-up appears, with options to create a diploid homozygous for the selected allele, or a heterozygous diploid with the wild type allele. If more than one allele has been entered for the gene, a heterozygous genotype will appear for each additional allele:

To create a multi-allele genotype, first add all of the constituent single alleles to the single-locus genotype table. Then select two or more alleles by ticking the boxes at the left side of the table. The "Combine selected genotypes" button becomes enabled:
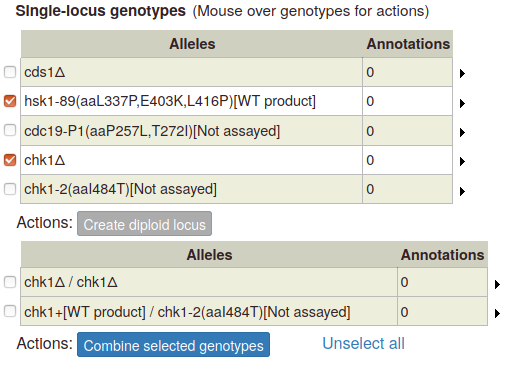
Click the button to add the new genotype to the table of multi-allele genotypes:
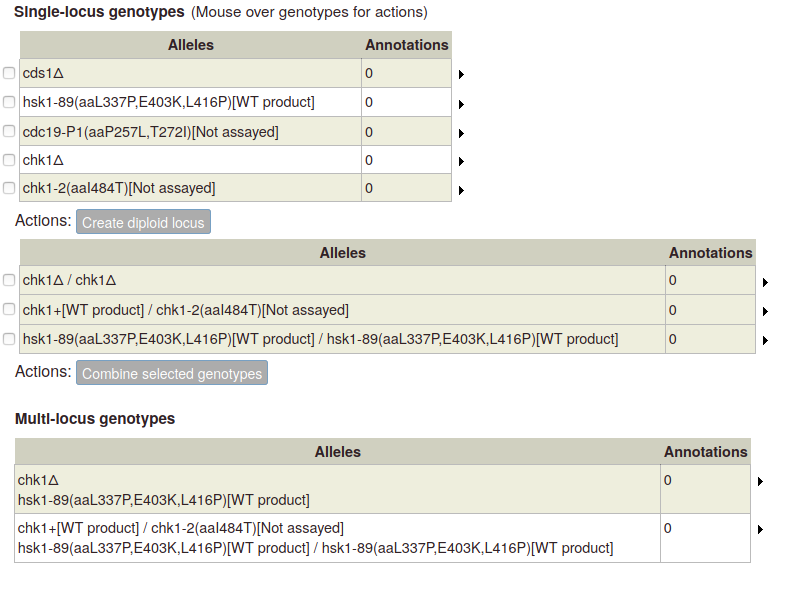
For multi-locus diploid genotypes, create single-locus diploid genotypes, which you can then select and combine.
Using wild-type alleles in genotypes: A wild type gene at its normal (endogenous) expression level should not have phenotypes annotated in Canto. Single wild-type alleles should only have phenotypes if the gene is expressed at a higher (overexpression) or lower (knockdown) level than normal. Similarly, in a multi-allele genotype, the wild-type allele is the only allele of the gene present, it should only be included if it is over- or underexpressed. Although it is possible to add a wild type as a single allele with wild type expression, this is intended only for use in cases where both wild-type and mutant alleles of the same gene are present (usually done to test whether the mutation is dominant over wild type). In these multi-allele genotypes the wild-type allele can have wild type expression specified.
Using, editing, deleting and duplicating genotypes
Mouseover options: When you mouse over any genotype in either table, a set of options appears in a popup:
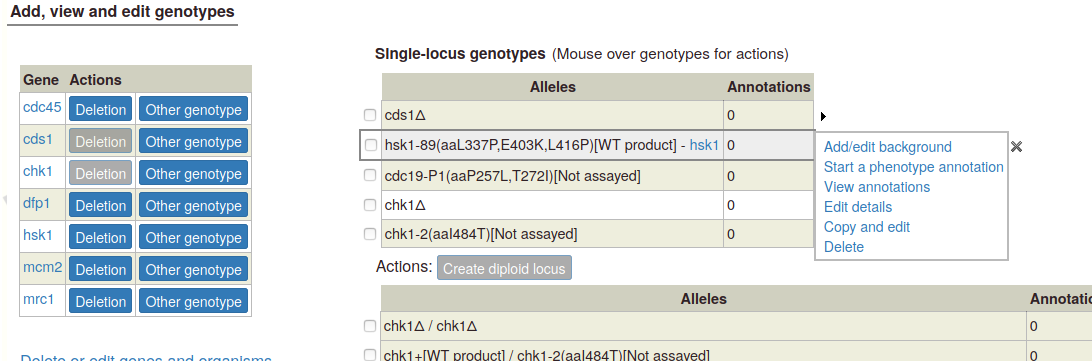
Add/edit background: This link pops up a text box where you can enter background alleles (we plan to add more sophisticated allele lookup later). If any background details are included, they appear in a column in the genotype table:
Note:If a single allele has a background, the background will be included with any multi-allele genotype that uses the allele. If two or more alleles have backgrounds, the backgrounds will be combined in the multi-allele genotype (duplicate alleles will only be included once). To change the background, use the one of the "Add/edit background", "Edit details", or "Copy and edit" options.
Start a phenotype annotation: The phenotype annotation procedure is identical to the single-allele curation interface -- find a suitable FYPO term, enter evidence and conditions, and finish the annotation all as described above. You can return to the Genotypes page at any time to select a genotype and add a phenotype for it, or to add or edit genotypes.
View annotations: This links to a page that lists the genotype details, a table of phenotypes annotated for the genotype, and an additional link to the phenotype annotation interface.
Edit details: On the genotype editing page, you can edit the details (name, description, expression) of alleles in the genotype, remove alleles, or add text describing the background. You can also add alleles using the gene table on the left, but this will usually be slower than combining single alleles on the genotype management page. Note that changes will apply to any phenotypes already annotated to the genotype.
Copy and edit: This links to the genotype editing page as described above, but creates a new genotype with the amended details (and no annotations).
Delete: The "Delete" link will ask you to confirm that you want to remove a genotype, and then delete it. The link is disabled for any genotype that has phenotype annotations. To delete a genotype with annotations, first delete the annotations (use the "view annotations" link or return to the paper summary page).
Editing, deleting and duplicating phenotypes
Edit: If you want to make changes to an annotation you have made, use the "Edit" link next to the annotation in the table. In the pop-up edit the appropriate fields, then click "OK".
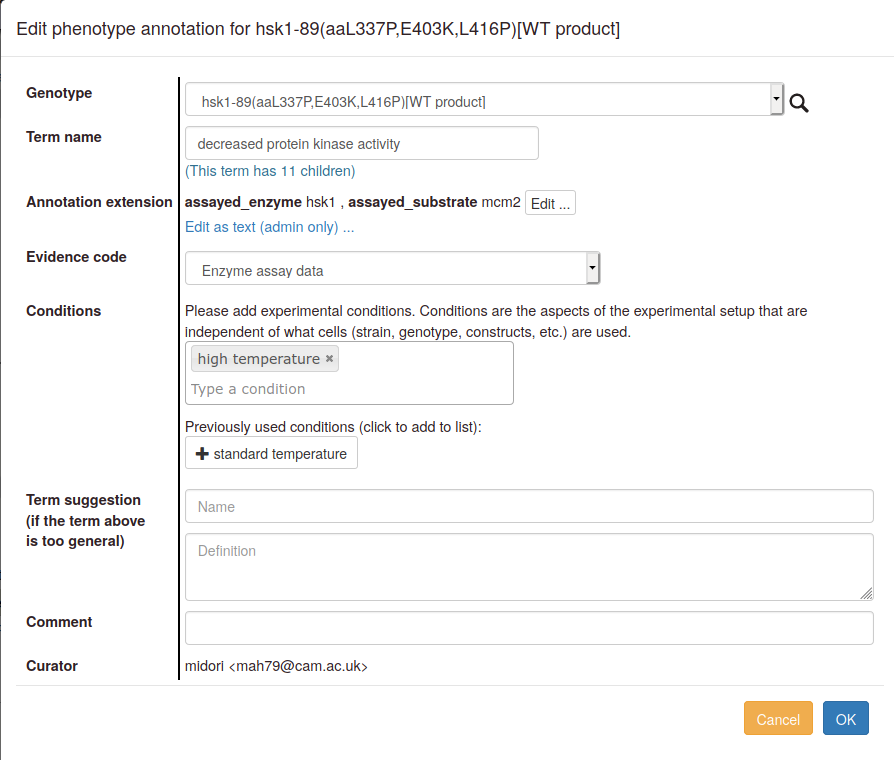
Delete: The "Delete" link will ask you to confirm that you want to remove an annotation, and then delete it.
Copy and edit: The "Copy and edit" link allows you to make a new annotation to the same allele or genotype, or transfer the annotation any other genotype in the genotype list (with or without changing any other details). For example, you may want to indicate that you have observed a phenotype under more than one set of conditions, e.g. at both standard and high temperatures. The interface works the same way as for editing an annotation, except that a new annotation is created, and the old annotation is retained without changes.
Quick Add: The "quick add" links available in advanced mode open the editing pop-up without any data entered or selected. Note that you cannot create alleles or genotypes in the "quick" phenotype interface; it uses genotypes that have already been entered in the Canto session.
Annotation extensions
You can add annotation extensions to provide additional specificity for FYPO annotations. (See the PomBase documentation for more information on annotation extensions.) After you have selected an ontology term and evidence, the Canto interface will display any available extension types. Click the link to choose an extension type and bring up a pop-up in which you specify the required details for the extension. For example, an annotation to "protein mislocalized to nucleus" can have any of these extensions:
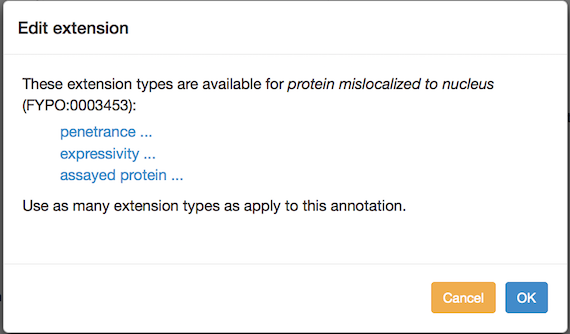
If more than one type is offered, you can add one of each type, but if you add them at the same time they will be interpreted as going together to form a compound annotation in which all of the parts apply at once. To create independent annotations (i.e. where one or another may apply, but not necessarily all at once), finish the annotation with one extension (or set of extensions), and then use the "Copy and edit" feature to create another annotation where you can edit the extension(s).
In all cases, the actual relation name used by the database will appear when you have finished the annotation plus extensions.
When you edit or duplicate an annotation, extensions can also be added, amended or deleted. An "Edit" button in the pop-up launches the annotation extension addition steps.

To change an existing extension, first delete it and then add a new one. Editing interface ("protein mislocalized to nucleus" example):
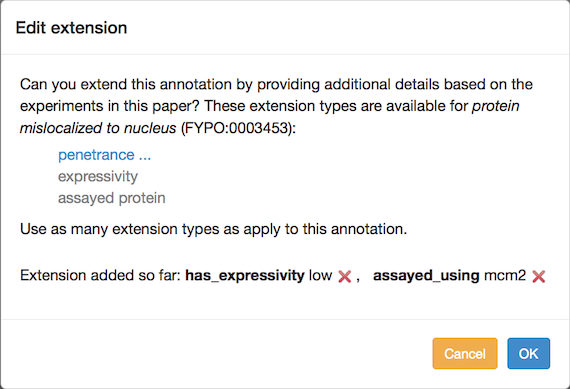
Phenotype annotation extensions can be used to indicate:
- Penetrance: the proportion of a population that shows the phenotype. The penetrance measurement can be qualitative or quantitative. The pulldown menu for qualitative options is selected by default. For a quantitative value, switch the radio button and enter a percentage (e.g. 38%) in the text box.
- Severity: Only qualitative values are supported; choose from the pulldown menu. (Note: severity was previously called "expressivity", and can still be used in the sense of the extent to which a phenotype is expressed.)
- A specific gene, RNA or protein, used in an assay (see "Targets of mutated genes" and "Localization phenotypes" below). The pulldown menu is populated with genes from the list you entered for the paper. You can add another gene at this point if necessary. For some phenotypes, a Sequence Ontology (SO) term can be selected to describe a more general sequence region or class of transcripts. In these cases, the ontology-search autocomplete box appears, and searches SO. Type text, choose from the suggestions, and drill down to more specific terms as usual.
More details on annotation extension options are available in the "phenotype tips" sections below.
General phenotype tips
Phenotype vs. GO annotations
GO annotations should always reflect a gene's direct involvement in, or role in regulating, processes or functions. In contrast, phenotype annotations indicate that a mutation causes a change in a process, but may reflect downstream or indirect effects.
For example, cells mutated for a gene involved in cell wall biosynthesis may display defects in cytokinesis. This gene should not be annotated to the GO process term "cytokinesis" because its effect on cytokinesis is an indirect effect of improper cell wall biosynthesis, but the mutant allele may be annotated to the FYPO term "abnormal cytokinesis".
Most commonly, more phenotypes than GO terms can be extracted from a paper. Sometimes, several phenotype annotations but no GO annotations can be made, because the experiments do not definitively resolve the exact process that the gene is involved in.
Phenotype categories
Phenotypes may be observed at the level of a cell or its parts (e.g. "elongated cell", "abnormal cytokinesis", "abnormal microtubule cytoskeleton morphology"), or at a population level (e.g. "flocculating cells"). In particular, note that viability can be described at either the cell or population level, or both (also see below). Phenotypes affecting binding or enzymatic activities, such as cases where a mutation decreases enzymatic activity, can also be annotated. Phenotypes affecting molecular functions will be displayed with the "cell phenotypes" on PomBase gene pages (even if the activity was assayed in vitro).
Normal phenotypes
Often, a mutant will be assayed for a phenotype but will appear identical to wild-type cells in the assay. One or more of the "normal phenotype" terms, which are defined as having no observable difference from wild type under the given conditions, can be used to annotate these mutants. For example, an allele may be annotated to "normal growth on amphotericin B" or "normal cell morphology". A mutant may be "normal" in one respect, but abnormal in another (for instance, cells may grow at a normal rate, but have an abnormal morphology). Note that it is not necessary to annotate all respects in which a mutant cell is normal, only those that are unexpected or otherwise interesting.
Targets of mutated genes
For any phenotype where an effect on the amount, stability, localization, etc. of a specific gene or gene product (protein or RNA) was measured, an extension should be included to identify the genes, RNAs, or proteins used in the experiment. The reasons are that the measured product may be that of a gene that was not mutated, and that the mutation may affect different gene products differently. For example, deletion of gene A may result in an increased amount of RNA transcribed from gene B; or a point mutation in A may cause decreased phosphorylation of protein B but not protein C. In these cases the annotation should be made to gene A, and an extension should be included that gene B is affected (and that C is not affected, in the second example). Sometimes it may be necessary to specify two targets. For instance, if a mutation in gene A cause "decreased binding" between protein B and protein C, include both B and C in the comment.
Localization phenotypes
If the localization of a protein or RNA is altered in a mutant compared to wild type, this should be annotated as a phenotype of the mutated gene (which may be different from the gene encoding the mislocalized product). Include an extension to identify the mislocalized protein/RNA, as described above. For example, if deletion of gene A causes gene B to localize to the cytoplasm instead of the nucleus, annotate A to an "abnormal protein localization" term (such as "abolished protein localization to nucleus, with protein mislocalized to cytoplasm"), and mention B in an extension.
Phenotypes of wild-type cells
Please note that only mutant phenotypes should be annotated in Canto -- i.e., the characteristics of cells in which DNA sequence of a gene is altered, or when its expression level is changed (or both). Any changes occurring in wild-type cells in response to a stimulus (such as an effect resulting from the inhibition of a wild-type gene when it binds a substance) can therefore not be captured as a phenotype. Please include information of this sort in a comment, and PomBase curators will advise you how to proceed (for example, it may be possible to use a GO annotation with an extension).
Specific phenotype tips
Cell and population viability
To capture whether a mutant is viable or inviable overall, use one of the cell population viability terms. As noted in the PomBase FAQ on essential genes, the most commonly used terms are "viable vegetative cell population" (FYPO:0002060) and "inviable vegetative cell population" (FYPO:0002061).
To describe characteristics of viable or inviable cells, FYPO has a wide selection of "viable cell" and "inviable cell" terms that also specify other features such as shape, presence and number of nuclei or septa, etc. If one mutation gives rise to a mixed population, more than one term may be used for the same allele, including both "viable" and "inviable" terms, and extensions should be used to indicate penetrance. See the relevant FAQ for more information.
Cell growth vs. cell population growth
Most experiments that measure "growth" observe a population of cells, such as a culture in liquid medium or a colony on a plate. For these experiments, use one or more of the "cell population growth" terms. In contrast, terms such as "normal cell growth" refer specifically to growth in the sense of an increase in cell size. Also see "Slow growth, decreased cell density and decreased growth" below.
Slow growth, decreased cell density and decreased growth
"Slow cell population growth" refers specifically to when the rate of growth (slope) of a cell population is decreased. "Decreased cell density in stationary phase" refers to when a population reaches stationary phase at, and maintains, a lower cell density than wild type (the rate of growth may still be the same as wild-type). An allele may be annotated to both terms. If you do not know which of these is decreased (typically if cells are grown on agar plates for a number of days and then observed) use "decreased cell population growth".
Cellular structures vs. processes affecting cellular structures
FYPO includes many pairs of terms that refer to the same cellular or subcellular structure, where one describes the structure (as normal, abnormal, etc.) and the other describes a process affecting the structure. In each pair, the structural phenotype term uses a GO cellular component (CC) term in its logical definition, and the process phenotype term uses a GO biological process (BP) term. The terms will often be linked by a has_output relation. For example, "abnormal kinetochore morphology" (FYPO:0000050) refers to the GO CC term "kinetochore" (GO:0000776), and "abnormal kinetochore organization" (FYPO:0000807) refers to the GO BP term "kinetochore organization" (GO:0051383).
Use the process phenotype term to annotate experiments in which the assay can monitor the process as it occurs (e.g. if you have movies, kymographs, a reconstituted system, or even a sufficiently detailed series of still pictures). Otherwise, use the structure or morphology phenotype terms (e.g. when you have only one or a few still pictures).
Translation vs. protein level
Typically, assays such as western blots do not distinguish between increased translation or decreased degradation. If these cannot be distinguished between then the correct terms to use are increased, decreased, or normal protein level. If the assay is specific enough, then the translation or degradation terms may be used (e.g. "decreased translation" or "increased protein degradation"). In either case, the specific protein(s) assayed can be put in annotation extensions.
Transcription vs. RNA level
Typically, assays such as northern blots do not distinguish between increased transcription or decreased degradation. If these cannot be distinguished between then the correct terms to use are increased, decreased, or normal RNA level. If the assay is specific enough, then the transcription or degradation terms may be used (e.g. a run-on assay can support annotation to "decreased transcription"). In either case, the specific gene(s) assayed can be put in annotation extensions.
Septation
Usually, "abnormal septum assembly" (FYPO:0000117), "normal septum assembly" (FYPO:0000673), or one of the more specific "septum assembly" terms will apply. Also consider annotating to a "actomyosin contractile ring contraction" term (abnormal, FYPO:0001364; normal, FYPO:0004097; or a more specific term) in cases where "septation" is used to refer to the coordinated processes of ring contraction and septum formation.